Project Partner:
Tobias Kind, Kwang-Hyeon Liu, Do Yup Lee, Brian DeFelice, John K. Meissen, Oliver Fiehn
Results:
LipidBlast in silico tandem mass spectrometry database for lipid identification
Published in Nature Methods (2013); Advanced Online; LINK: doi:10.1038/nmeth.2551
PubMed Central Link (Open Access): PMC3731409
Short Introduction:
Lipid analysis of polar lipids can be performed with tandem mass spectrometry and mass spectral library search. We developed a computer-generated (in-silico) tandem mass spectral (MS/MS) database that can be used to annotate and identify hundreds of lipids in plants, bacteria, algae, animals, humans, viruses. Our LipidBlast library works with low-resolution and high-resolution instruments. See also our LipidAnalysis web page [LINK].

Lipid-Classes -this link details about the covered classes and number of spectra in LipidBlast
LipidBlast-Templates - this explains how to study and create lipid fragmentations using EXCEL
LipidBlast-mz-lookup - explains how to perform an MS1 search only and discusses the needed resolving power
MGF-files -explains how to create MGF files that contain all MS/MS scans and precursor ion information
MS/MS-GUI-search - details how to search MS/MS spectra in visual mode using the NIST MS Search GUI
MS/MS-batch-search - explains how to annotate thousands of spectra in a few seconds with EXCEL output
Download LipidBlast:
The latest LipidBlast tools have been integrated into MS-Dial software. [LINK]
Picture service (for your convenience):
Parts of the software supplement of the publication are published under the Creative Commons (by) license. This license lets others distribute, remix, tweak, and build upon your work, even commercially, as long as they credit you for the original creation. Download PPT and TIFF pictures (please see ZIP above).
Publications and presentations using LipidBlast:
Daily updated citation statistics from Google Scholar [LINK]
- Induced Pluripotent Stem Cells Show Metabolomic Differences to Embryonic Stem Cells in Polyunsaturated Phosphatidylcholines and Primary Metabolism; PLOS ONE 2012 [LINK]
- Qualitative analysis of algal secretions with multiple mass spectrometric platforms;
Journal of Chromatography A; Volume 1244, 29 June 2012, Pages 139–147 [LINK] - Analysis of Polar Lipids in Chlamydomonas reinhardtii Using Nanoelectrospray Direct Infusion Method and Gas Chromatography and Mass Spectrometric Detection; ACTA CHIMICA SINICA 2013 [LINK]
- Lipid identification using a MS/MS database of 120,000 tandem mass spectra; (Poster);
ASMS 2010 - 58th ASMS Conference on Mass Spectrometry; Salt Lake City, Utah [PDF] - De novo identification of small molecules with computer generated MS/MS libraries; (Poster);
ASMS 2011 - 59th ASMS Conference on Mass Spectrometry; Denver, Colorado [PDF] - Generation of in-silico MS/MS mass spectra using combinatorial algorithms and reaction prediction expert systems; (Invited Talk); American Chemical Society - National Meeting 239, San Francisco, Spring, 2010 [PDF]
- Lipid identification by tandem mass spectrometry using an in-silico generated fragment database; (Poster); Metabolomics Society Conference 2009 - Edmonton, Alberta, Canada [PDF]
- HIGH THROUGHPUT IMPLEMENTATION OF REVERSED-PHASE UHPLC–CSH-QTOF MS/MS FOR COMPREHENSIVE ANALYSIS OF COMPLEX LIPIDS IN BLOOD PLASMA (P11-28); 9TH ANNUAL CONFERENCE OF THE METABOLOMICS SOCIETY; Glasgow 2013
- A novel UHPLC-Q-TOF method for the detection and quantification of plasma lipids; ASMS Conference Minneapolis, Minnesota TP 480 [PDF]
- COMPARING DIFFERENT ANTICOAGULANTS FOR SAMPLING BLOOD PLASMA USING GC-TOF-MS AND LC-QTOF-MS/MS METABOLOMICS (P5-16); 9TH ANNUAL CONFERENCE OF THE METABOLOMICS SOCIETY; Glasgow 2013
- OPTIMIZING SAMPLE PREPARATION FOR METABOLOMICS COVERAGE IN ROBUSTNESS IN YEAST (P10-4); 9TH ANNUAL CONFERENCE OF THE METABOLOMICS SOCIETY; Glasgow 2013
- Polar lipid profiling of Euglena gracilis using LipidBlast In-silico MS / MS library
Japan Society for Bioscience Conference Abstracts (Web) Volume: 2013 : page 3B11A10 [LINK] - Respiration Metabolism During Thermoinhibition of Lettuce (Lactuca sativa) Seeds;
American Society of Plant Biologists - Western Section 2013 Meeting - How can we get data for “all metabolites” in Chlamydomonas reinhardtii ?
Conference on Small Molecule Science (CoSMoS) Meeting San Diego 2013 - Pitfalls in long-term and large-scale LC-MS-based lipidomics studies of human plasma
62nd ASMS Conference on Mass Spectrometry and Allied Topics, June 15 - 19, 2014
Links to external software used in the project:
Programs:
- TEXTPAD ($$) www.textpad.com
- MS EXCEL ($$$) + Visual Basic www.microsoft.com
- ChemAxon molconvert (free), cxcalc (academic license), JCHEM full (academic license)
- ChemAxon Instant-JChem (free academic version)
- LipidMaps Tools
- SMILIB from ModLab Frankfurt
- NIST MS Search- for manual inspection of MS/MS spectra
- NIST MS PepSearch - for batch processing of millions of spectra
- ProteoWizard - for creation of MGF files from all vendor MS data

NIST GUI for visual inspection
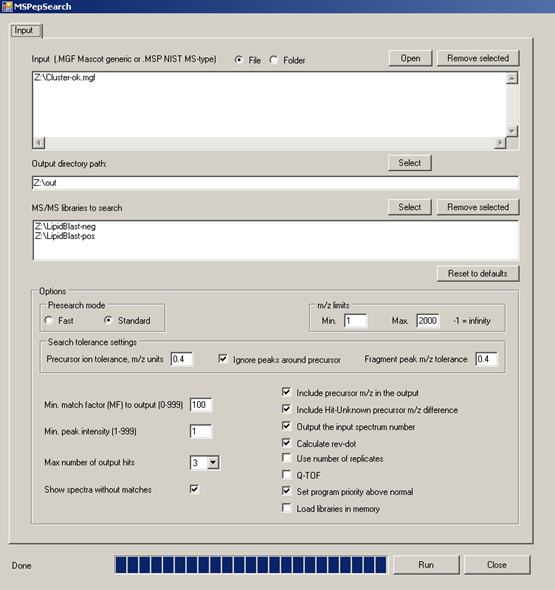
MSPepSearch BATCH GUI for high-throughput processing of MGF files and EXCEL output of hit scores
Databases and Services (updated):
- The PubChem database (free) - download the whole PubChem DB here: PubChem FTP
- LipidMaps database (free)
Frequently Asked Questions (FAQ):
- Q: Is LipidBlast right for me?
A: YES, its so simple a cave-men could do it. However I recommend to use also other tools from my lipid analysis collection page [LINK]. LipidBlast integrated into NIST MS Search (an independent program) is a natural way for spectral search and visual inspection, compared to other black box programs. - Q: Why are there no structures in the database included?
A: They actually are, they can be found in the development sheet or externally. At the time of development the structure files were several hundred MByte large, so the userbase would have been limited. I opted for a smaller and faster database. - Q: I have an UPLC-MS/MS experiment, but get many so duplicates hits.
A: If you have a fast-scanning MS/MS instrument that will happen a lot, but thats not a problem with LipidBlast per se. I recommend a post-processing option that excludes those duplicates. - 4) Q: Is LipidBlast for Windows only or for MACs or LINUX? A: LipidBLast can be used on all platforms, see the HandBook.
- Q: Why are there no InchiKeys integrated?
A: Because when LipidBlast development started the InchiKey was not released yet. However they can be created with the development sheet and other structures. - Q: How can I do batches of hundreds of files?
A: Use NIST MS Pepsearch (see above) and refer to the handbook (see above) it explains how to create score reports that can be opened in EXCEL. Its quite fast, search speeds of 1000 spectra/second can be reached. - Q: Is there an online service like MassBank, METLIN, MetaboAnalyst?
A: NO, but the ASCII libraries can be imported into any search service even online, so its not a problem with LipidBlast per se. - Q: How can I quantitate or quantify lipids.
A: This can be done with any vendor specific MS program (use peak integration). For large datasets use alignment programs, see here [LINK]. - Q: How many lipids can I identify in an average experiment?
A: Depends on the organism, for plants, algae, animals minimum 50 to 250. See our paper in Nature Methods. - Q: Is LipidBlast for GC-MS?
A: No. Only for LC-MS/MS and LC-MS data. - Q: There are lots of lipids missing, why?
A: Yes, thats why we created half of the structures in LipidBlast using combinatorial enumeration. Still many classes are missing, because not enough spectral data and fragmentation data could be found in older publications or databases. - Q: I only have an LC-MS instrument, can I use LipidBlast?
A: YES, but only if its a accurate mass instrument, or software calibrated to generate accurate masses. Examples are LC-TOF-MS, Citius LC-HRT or Orbitrap Exactive or any other vendor instrument. The "trick" is to use accurate mass lookup only. LipidBlast provides an m/z lookup table with all lipids and all adducts and accurate masses. The file LIPIDBLAST-mz-lookup.xls can be found in the download section. Be aware that isobars give multiple hits, so retention times should be included. - Q: I only have an older low-res ion trap, can I use LipidBlast?
A: Certainly, any low resolution or unit-mass instrument with MS/MS capability is useful, best in use with LC together. The limitation of MS/MS search in the past was that actually no large and diverse MS/MS databases existed! - Q: Which instrument vendors are compatible with LipidBlast?
A: Any, as long as they have MS/MS instruments and I found example spectra. We have examples from ABI Sciex, Agilent, Bruker, JEOL, Kratos, SHIMADZU, Thermo and Waters. There is a a list of more than hundred examples from different platforms in the Nature Methods supplement and the download section above, including ABI 4000 Q-Trap, ABI 4700 MALDI TOF/TOF, ABI 4800 MALDI-TOF/TOF, ABI API 2000 triple quadrupole, ABI QSTAR-XL QTOF, ABI QTRAP 4000, ABI Sciex API III QQQ, ABI-QSTAR-Pulsar-Quadrupol-TOF, ABI-QSTAR-XL-Quadrupol-TOF, Agilent 6410 triple quadrupole MS, Agilent 6520 Q-TOF, Agilent 6530 QqTOF, Agilent Ion Trap SL, Agilent Ion Trap XCT, Agilent LC/MSD 1100 Ion Trap , Agilent MSD 1100 single quadrupole MS, Agilent QTOF, Bruker DESI FTICR APEX-Q, Bruker Esquire 3000 ion trap, Bruker Esquire ion trap, Bruker UltraFlex II MALDI TOF-TOF, Bruker microTOF qQ-TOF, Bruker ultrafleXtreme MALDI TOF/TOF, JEOL JMS-HX110A/110A Tandem MS FAB, Kratos MALDI-TOF AXIMA-CFR, SHIMADZU KRATOS MALDI TRAP TOF, SHIMADZU LCMS-IT-TOF, Thermo Finnigan DecaXP ion trap, Thermo Finnigan LCQ DECA ion trap, Thermo Finnigan LTQ linear ion trap, Thermo Finnigan TSQ Quantum Triple Quadrupole, Thermo Fisher Exactive Orbitrap, Thermo Fisher LTQ-FT, Thermo LTQ Orbitrap, Thermo LXQ iontrap, Thermo Orbitrap Velos ESI , Waters AutoSpec magnetic sector MS, Waters MicroMass QqQ triple quadrupole, Waters Micromass Q-TOF Micro , Waters QTOF Premier, Waters QqQ triple quadropole VG Quattro II, Waters Synapt HDMS, Waters micro QTOF. - Q: What is the best way to acquire MS/MS spectra?
A: On fast scanning instruments (QTOF or Orbitraps and others) the best way is to allow for multiple CID or HCD fragmentation voltages such as 0V, 20V, 40V, 60V. In that way fragmentation rich tandem mass spectra are created and hit/match factors are improved. - Q: Profile mode or centroid mode, what is better?
A: For spectral searching and matching it is important to centroid the MS/MS spectra before searching (hence create stick spectra). Additionally the number of product ions in the spectrum should be reduced to less than 100. That will tremendously improve the search speed and in some cases also improve hit quality. The reasons are that matching algorithms include the m/z and abundance pairs of the whole spectrum. Controiding is best performed in the platform software or if needed with ProteoWizard MSConvert (peak picking).